This function calculates (conditional-)reciprocal best hit
(CRBHit) pairs from two CDS DNAStringSet's.
CRBHit pairs were introduced by Aubry S, Kelly S et al. (2014).
Sequence searches are performed with last
Kiełbasa, SM et al. (2011) [default]
or with mmseqs2
Steinegger, M and Soeding, J (2017)
or with diamond
Buchfink, B et al. (2021).
If one specifies cds1 and cds2 as the same input a selfblast is conducted.
Usage
cds2rbh(
cds1,
cds2,
dbfile1 = NULL,
dbfile2 = NULL,
searchtool = "last",
lastpath = file.path(find.package("CRBHits"), "extdata", "last-1639", "bin"),
lastD = 1e+06,
lastm = 10,
mmseqs2path = NULL,
mmseqs2sensitivity = 5.7,
mmseqs2maxseqs = 300,
diamondpath = NULL,
diamondsensitivity = "--sensitive",
diamondmaxtargetseqs = 0,
lambda3path = NULL,
lambda3sensitivity = "sensitive",
lambda3nummatches = 25,
outpath = "/tmp",
crbh = TRUE,
keepSingleDirection = FALSE,
eval = 0.001,
qcov = 0,
tcov = 0,
pident = 0,
alnlen = 0,
rost1999 = FALSE,
filter = NULL,
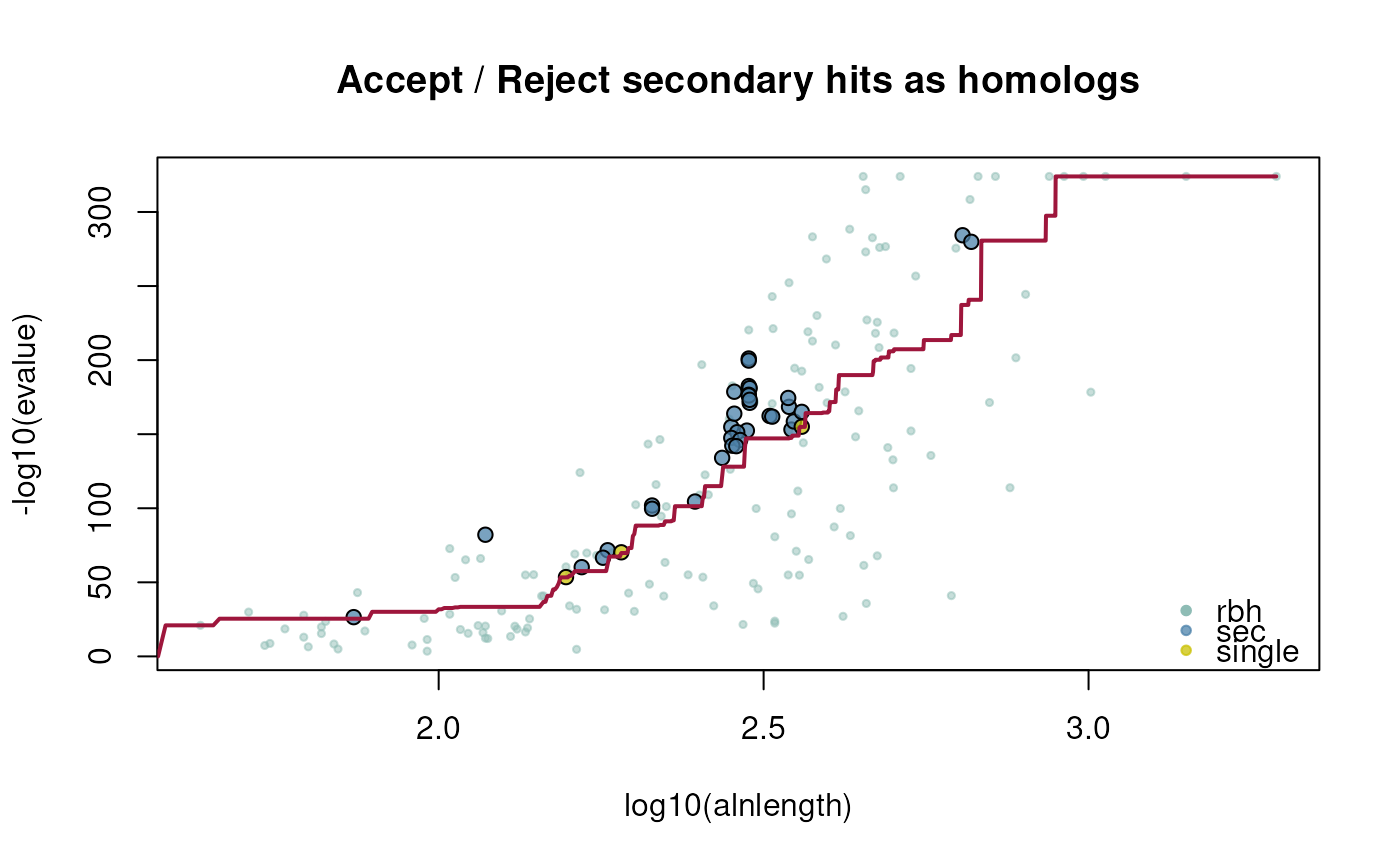
plotCurve = FALSE,
fit.type = "mean",
fit.varweight = 0.1,
fit.min = 5,
longest.isoform = FALSE,
isoform.source = "NCBI",
threads = 1,
remove = TRUE,
remove.db = TRUE,
shorten1 = FALSE,
frame1 = 1,
framelist1 = NULL,
genetic.code1 = NULL,
shorten2 = FALSE,
frame2 = 1,
framelist2 = NULL,
genetic.code2 = NULL
)Arguments
- cds1
cds1 sequences as
DNAStringSet[mandatory]- cds2
cds2 sequences as
DNAStringSet[mandatory]- dbfile1
aa1 db file [optional]
- dbfile2
aa2 db file [optional]
- searchtool
specify sequence search algorithm last, mmseqs2, diamond or lambda3 [default: last]
- lastpath
specify the PATH to the last binaries [default: /extdata/last-1639/bin/]
- lastD
last option D: query letters per random alignment [default: 1e6]
- lastm
last option m: maximum initial matches per query position [default: 10]
- mmseqs2path
specify the PATH to the mmseqs2 binaries [default: NULL]
- mmseqs2sensitivity
specify the sensitivity option of mmseqs2 [default: 5.7]
- mmseqs2maxseqs
mmseqs2 option: Maximum results per query sequence allowed to pass the prefilter [default: 300]
- diamondpath
specify the PATH to the diamond binaries [default: NULL]
- diamondsensitivity
specify the sensitivity option of diamond [default: –sensitive]
- diamondmaxtargetseqs
specify the maximum number of target sequences per query option of diamond [default: 0]
- lambda3path
specify the PATH to the lambda3 binaries [default: NULL]
- lambda3sensitivity
specify the sensitivity option of lambda3 [default: sensitive]
- lambda3nummatches
specify the number of matches per query option of lambda3 [default: 25]
- outpath
specify the output PATH [default: /tmp]
- crbh
specify if conditional-reciprocal hit pairs should be retained as secondary hits [default: TRUE]
- keepSingleDirection
specify if single direction secondary hit pairs should be retained [default: FALSE]
- eval
evalue [default: 1e-3]
- qcov
query coverage [default: 0.0]
- tcov
target coverage [default: 0.0]
- pident
percent identity [default: 0.0]
- alnlen
alignment length [default: 0.0]
- rost1999
specify if hit pairs should be filter by equation 2 of Rost 1999 [default: FALSE]
- filter
specify additional custom filters as list to be applied on hit pairs [default: NULL]
- plotCurve
specify if crbh fitting curve should be plotted [default: FALSE]
- fit.type
specify if mean or median should be used for fitting [default: mean]
- fit.varweight
factor for fitting function to consider neighborhood [default: 0.1]
- fit.min
specify minimum neighborhood alignment length [default: 5]
- longest.isoform
specify if cds sequences should be reduced to the longest isoform (only possible if data was accessed from NCBI or ENSEMBL) [default: FALSE]
- isoform.source
specify cds sequences source (either NCBI or ENSEMBL) [default: NCBI]
- threads
number of parallel threads [default: 1]
- remove
specify if last result files should be removed [default: TRUE]
- remove.db
specify if last db files should be removed [default: TRUE]
- shorten1
shorten all sequences to multiple of three [default: FALSE]
- frame1
indicates the first base of the first codon [default: 1]
- framelist1
supply vector of frames for each entry [default: NULL]
- genetic.code1
The genetic code to use for the translation of codons into Amino Acid letters [default: NULL]
- shorten2
shorten all sequences to multiple of three [default: FALSE]
- frame2
indicates the first base of the first codon [default: 1]
- framelist2
supply vector of frames for each entry [default: NULL]
- genetic.code2
The genetic code to use for the translation of codons into Amino Acid letters [default: NULL]
Value
List of three (crbh=FALSE)
1: $crbh.pairs
2: $crbh1 matrix; query > target
3: $crbh2 matrix; target > query
List of four (crbh=TRUE)
1: $crbh.pairs
2: $crbh1 matrix; query > target
3: $crbh2 matrix; target > query
4: $rbh1_rbh2_fit; evalue fitting function
References
Aubry S, Kelly S et al. (2014) Deep Evolutionary Comparison of Gene Expression Identifies Parallel Recruitment of Trans-Factors in Two Independent Origins of C4 Photosynthesis. PLOS Genetics, 10(6) e1004365.
Kiełbasa, SM et al. (2011) Adaptive seeds tame genomic sequence comparison. Genome research, 21(3), 487-493.
Rost B. (1999). Twilight zone of protein sequence alignments. Protein Engineering, 12(2), 85-94.
Examples
## compile last-1639 within CRBHits
CRBHits::make_last()
#> [1] "/__w/_temp/Library/CRBHits/extdata/last-1639/bin"
## load example sequence data
data("ath", package="CRBHits")
data("aly", package="CRBHits")
## get CRBHit pairs
ath_aly_crbh <- cds2rbh(
cds1=ath,
cds2=aly,
plotCurve=TRUE)
 dim(ath_aly_crbh$crbh.pairs)
#> [1] 211 3
## get classical reciprocal best hit (RBHit) pairs
ath_aly_rbh <- cds2rbh(
cds1=ath,
cds2=aly,
crbh=FALSE)
dim(ath_aly_rbh$crbh.pairs)
#> [1] 181 3
## filter for evalue 1e-100
ath_aly_crbh.eval100 <- cds2rbh(
cds1=ath,
cds2=aly,
eval=1e-100)
dim(ath_aly_crbh.eval100$crbh.pairs)
#> [1] 98 3
## filter for query coverage
ath_aly_crbh.qcov <- cds2rbh(
cds1=ath,
cds2=aly,
qcov=0.5)
dim(ath_aly_crbh.qcov$crbh.pairs)
#> [1] 145 3
## custom filter for e.g. bit score (column 12)
## Note: multiple filters can be given in filter param as list
myfilter <- function(rbh, value=500.0){
return(rbh[as.numeric(rbh[, 12])>=value, , drop=FALSE])
}
ath_aly_crbh.custom <- cds2rbh(
cds1=ath,
cds2=aly,
filter=list(myfilter))
dim(ath_aly_crbh.custom$crbh.pairs)
#> [1] 69 3
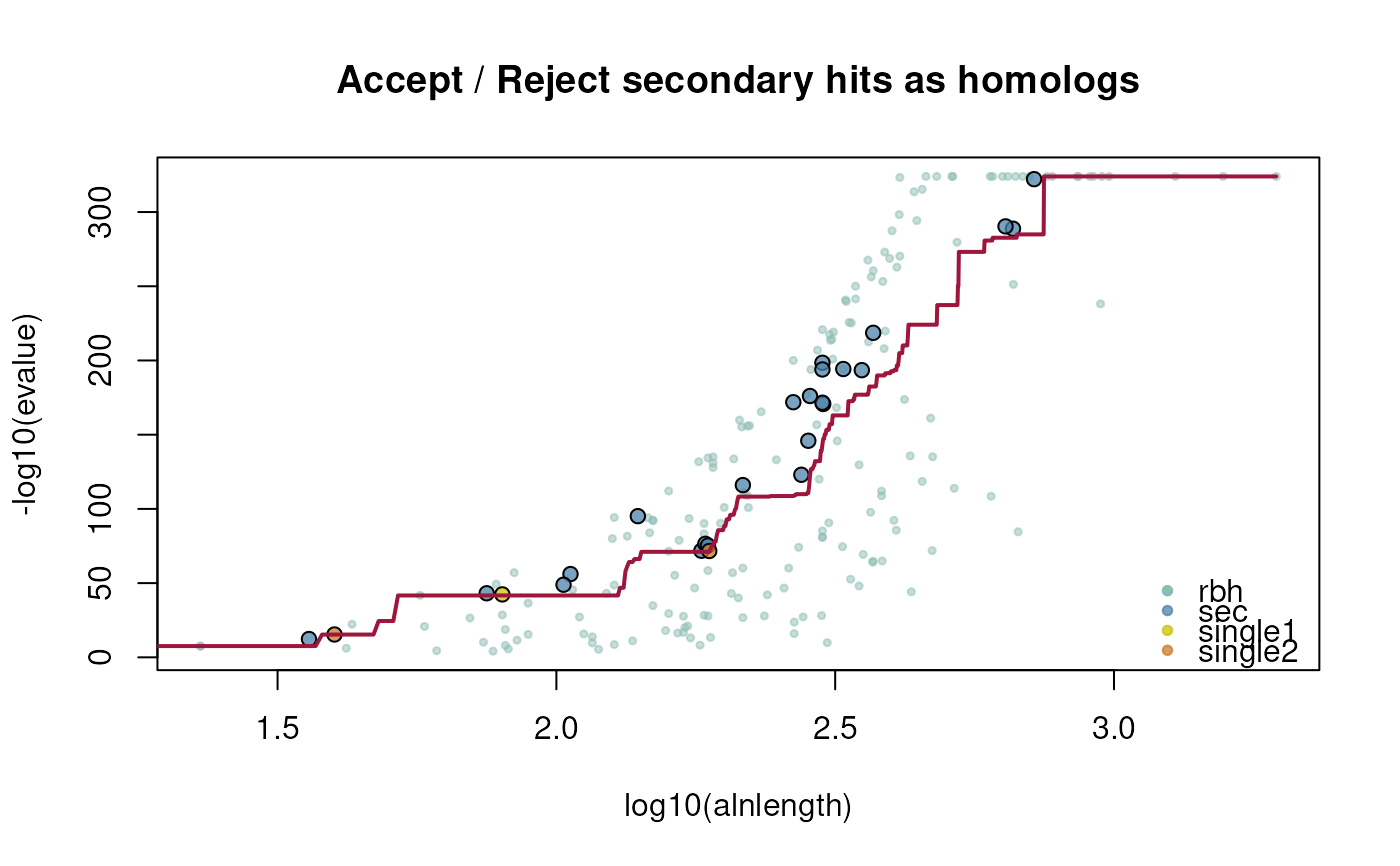
## selfblast
ath_selfblast_crbh <- cds2rbh(
cds1=ath,
cds2=ath,
plotCurve=TRUE)
dim(ath_aly_crbh$crbh.pairs)
#> [1] 211 3
## get classical reciprocal best hit (RBHit) pairs
ath_aly_rbh <- cds2rbh(
cds1=ath,
cds2=aly,
crbh=FALSE)
dim(ath_aly_rbh$crbh.pairs)
#> [1] 181 3
## filter for evalue 1e-100
ath_aly_crbh.eval100 <- cds2rbh(
cds1=ath,
cds2=aly,
eval=1e-100)
dim(ath_aly_crbh.eval100$crbh.pairs)
#> [1] 98 3
## filter for query coverage
ath_aly_crbh.qcov <- cds2rbh(
cds1=ath,
cds2=aly,
qcov=0.5)
dim(ath_aly_crbh.qcov$crbh.pairs)
#> [1] 145 3
## custom filter for e.g. bit score (column 12)
## Note: multiple filters can be given in filter param as list
myfilter <- function(rbh, value=500.0){
return(rbh[as.numeric(rbh[, 12])>=value, , drop=FALSE])
}
ath_aly_crbh.custom <- cds2rbh(
cds1=ath,
cds2=aly,
filter=list(myfilter))
dim(ath_aly_crbh.custom$crbh.pairs)
#> [1] 69 3
## selfblast
ath_selfblast_crbh <- cds2rbh(
cds1=ath,
cds2=ath,
plotCurve=TRUE)